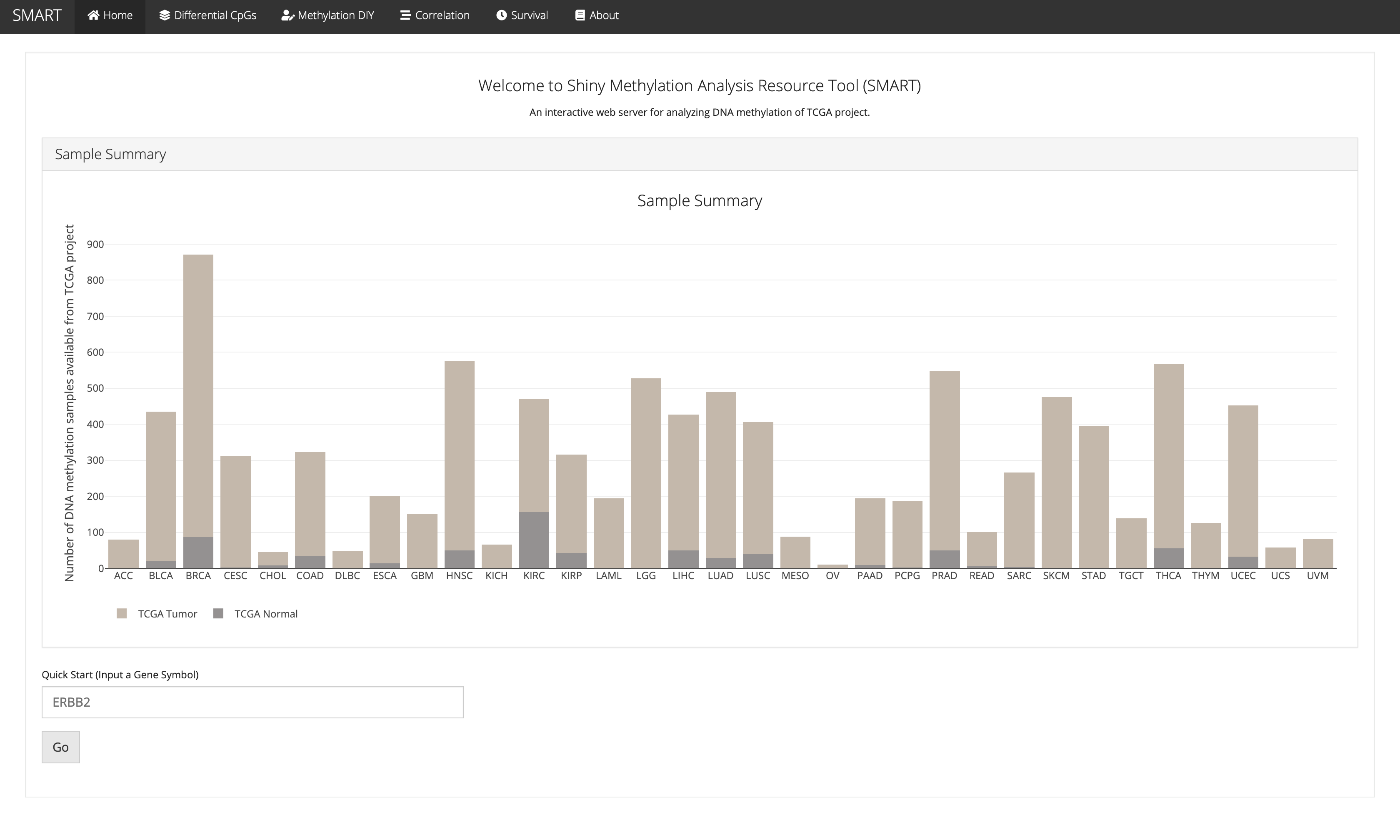
Welcome to Shiny Methylation Analysis Resource Tool (SMART)
Sample Summary
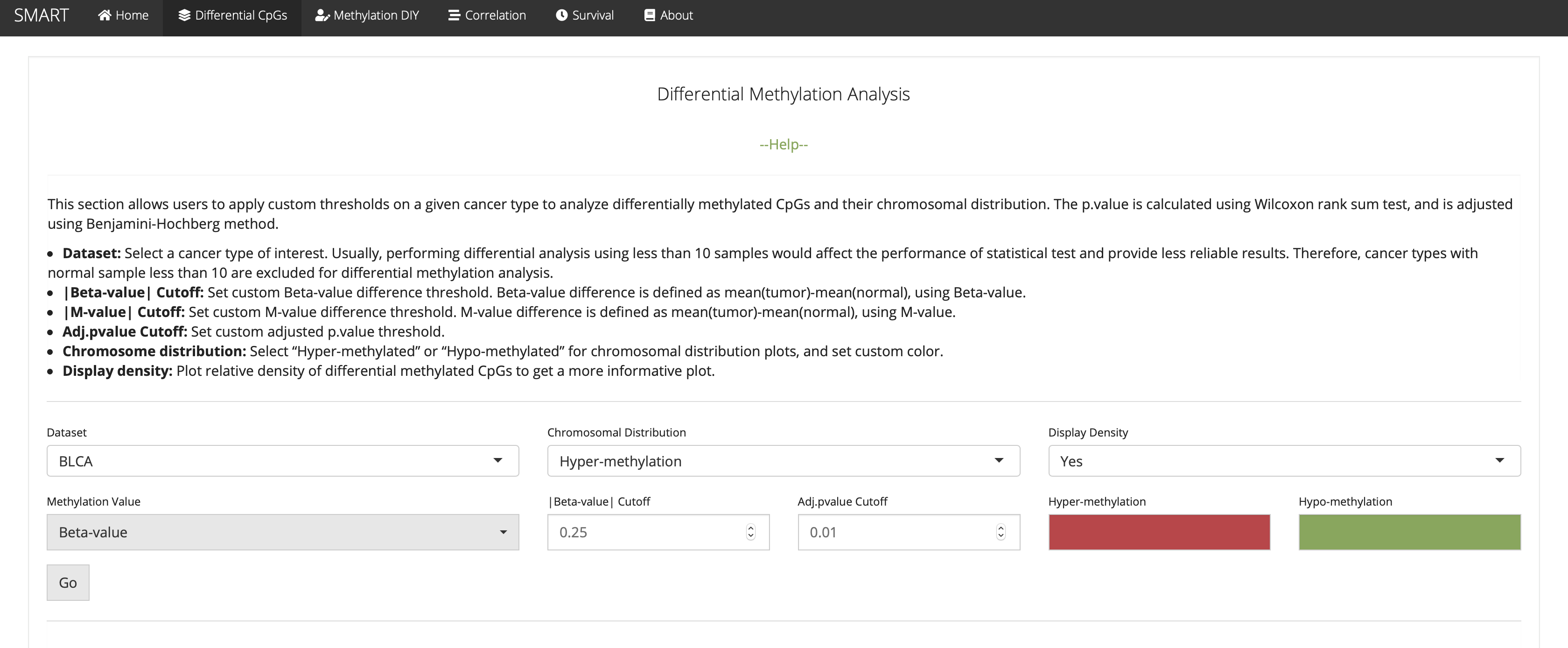
Differential Methylation Analysis
This section allows users to apply custom thresholds on a given cancer type to analyze differentially methylated CpGs and their chromosomal distribution. The p.value is calculated using Wilcoxon rank sum test, and is adjusted using Benjamini-Hochberg method.
Download
Differential methylated CpGs
Download
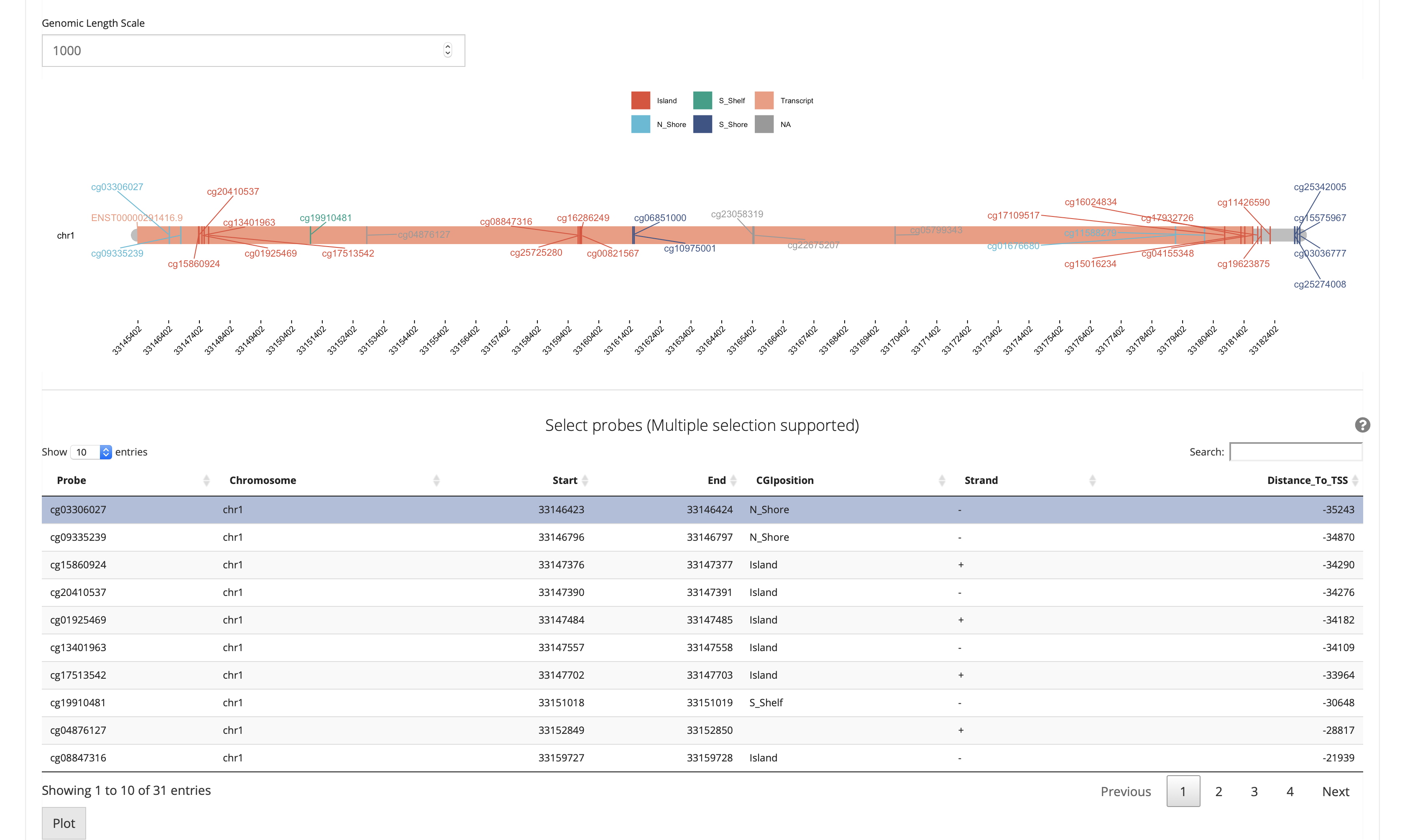
Sometimes users only want to visualize specific CpGs. To address this need, SMART offers a function that allows users to flexibly visualize CpGs. First, select CpGs that you want to plot from above table, check the corresponding chromosome locations. Next, select chromosomes that you checked from the above table in the panel, click the plot button, a beautiful circular plot showing the selected CpG sites and relative density will be displayed.
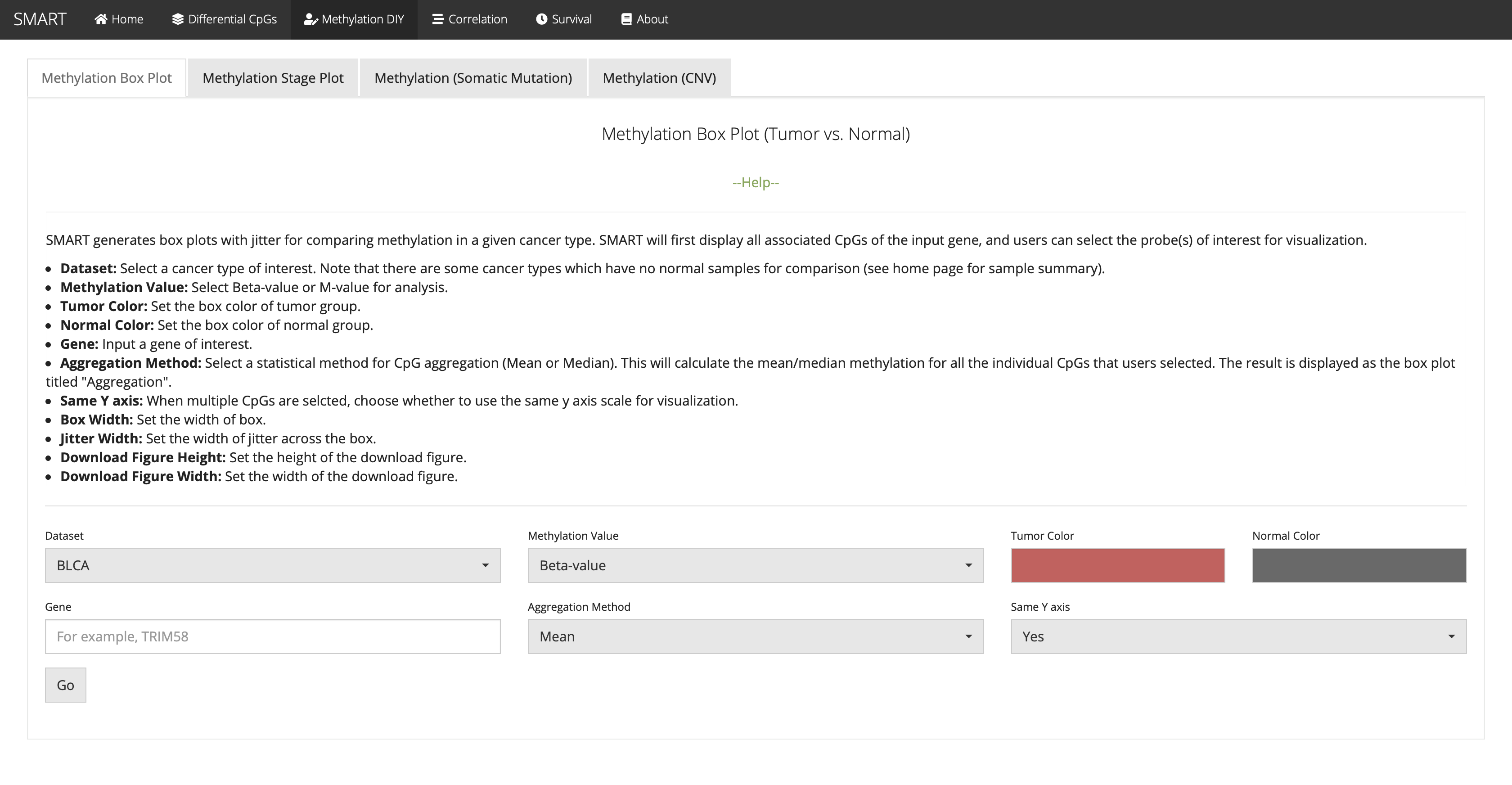
Methylation Box Plot (Tumor vs. Normal)
SMART generates box plots with jitter for comparing methylation in a given cancer type. SMART will first display all associated CpGs of the input gene, and users can select the probe(s) of interest for visualization.
Methylation Stage Plot
This feature displays methylation box plots based on the pathological stage.
Methylation (Somatic Mutation)
This feature allows users to compare methylation among tumors with or without the presence of mutation for a given gene.
Methylation (CNV)
This feature allows users to compare methylation among tumors with different copy number variations for a given gene.
Gene-level copy number variation (CNV) is estimated using the GISTIC2 threshold method.
The estimated values -2, -1, 0, 1, 2, representing homozygous deletion, single copy deletion, diploid normal copy, low-level copy number amplification, and high-level copy number amplification.
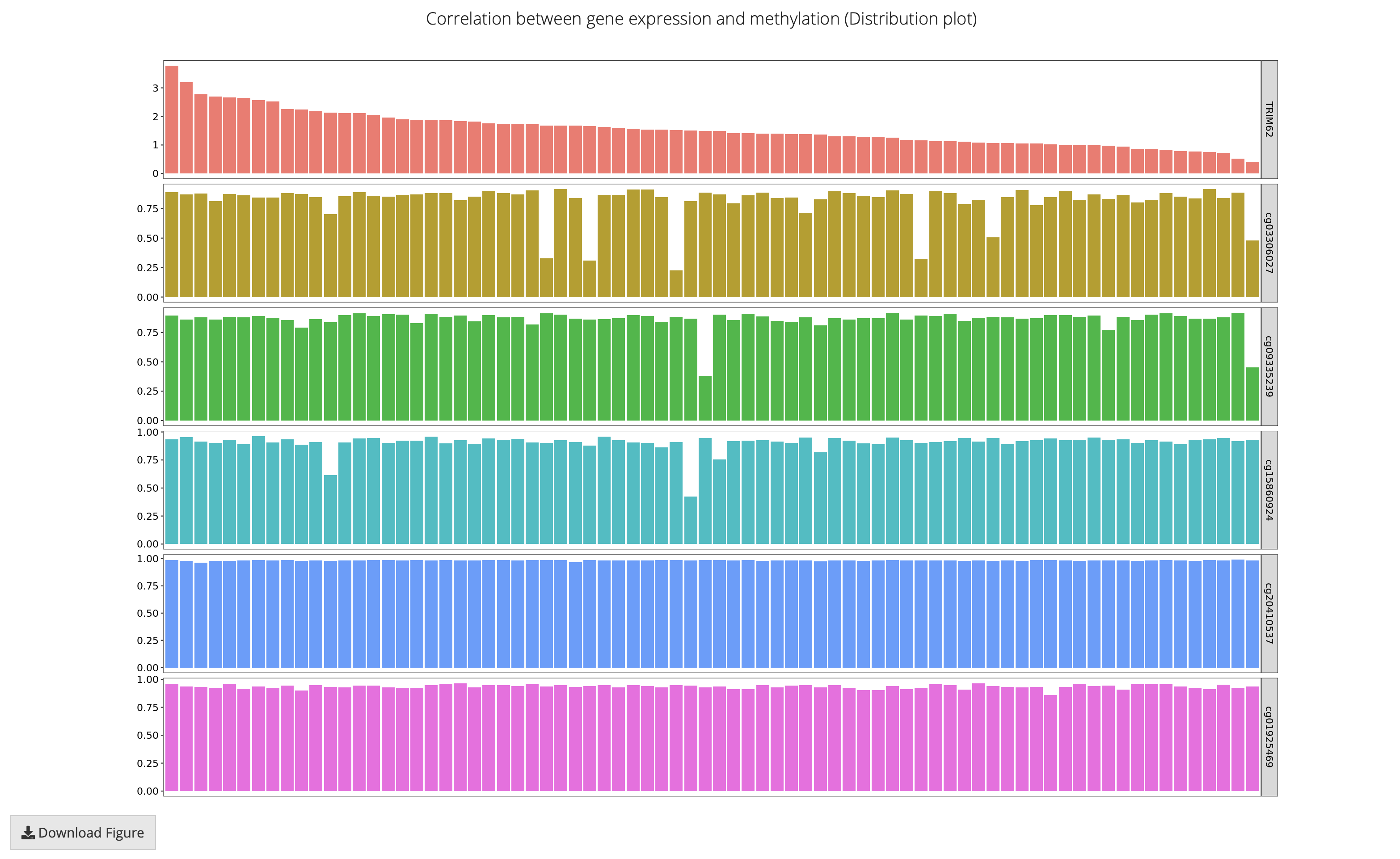
Correlation (Gene-level)
This feature performs pair-wise correlation analysis to explore the correlation between the expression and DNA methylation, using methods including Pearson, Spearman and Kendall. All samples, including normal samples (if available), with both gene expression and methylation data are included for correlation analysis.
SMART uses the log2-scaled(TPM+1) value (gene) and Beta-value or M-value (probe) for calculation.
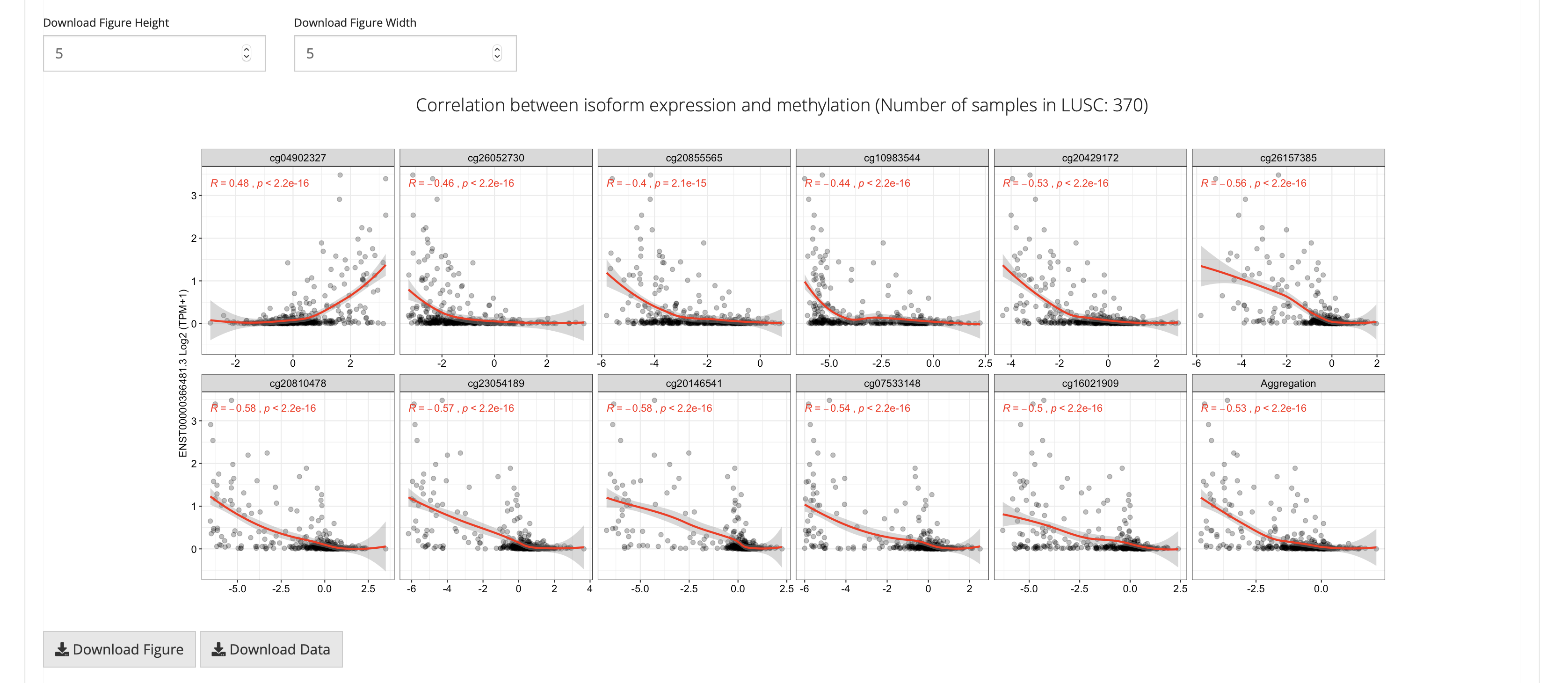
Correlation (Transcript-level)
This feature performs pair-wise correlation analysis to explore the correlation between the expression (transcript-level) and DNA methylation, using methods including Pearson, Spearman and Kendall.
SMART uses the log2-scaled(TPM+1) value (gene) and Beta-value or M-value (probe) for calculation.
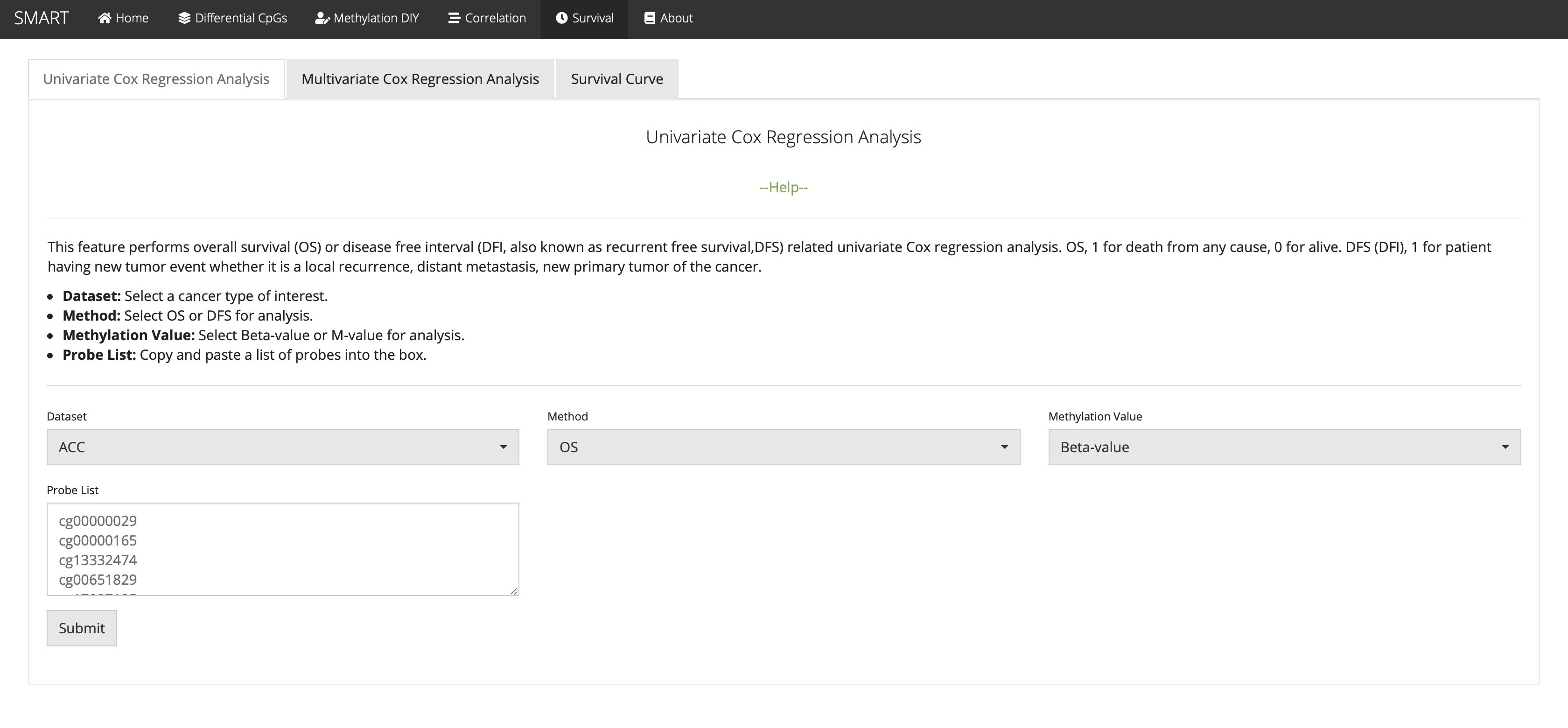
Univariate Cox Regression Analysis
This feature performs overall survival (OS) or disease free interval (DFI, also known as recurrent free survival,DFS) related univariate Cox regression analysis. OS, 1 for death from any cause, 0 for alive. DFS (DFI), 1 for patient having new tumor event whether it is a local recurrence, distant metastasis, new primary tumor of the cancer.
Multivariate Cox Regression Analysis
This feature performs overall survival (OS) or disease free interval (DFI, also known as recurrent free survival,DFS) related Multivariate Cox regression analysis. OS, 1 for death from any cause, 0 for alive. DFS (DFI), 1 for patient having new tumor event whether it is a local recurrence, distant metastasis, new primary tumor of the cancer.
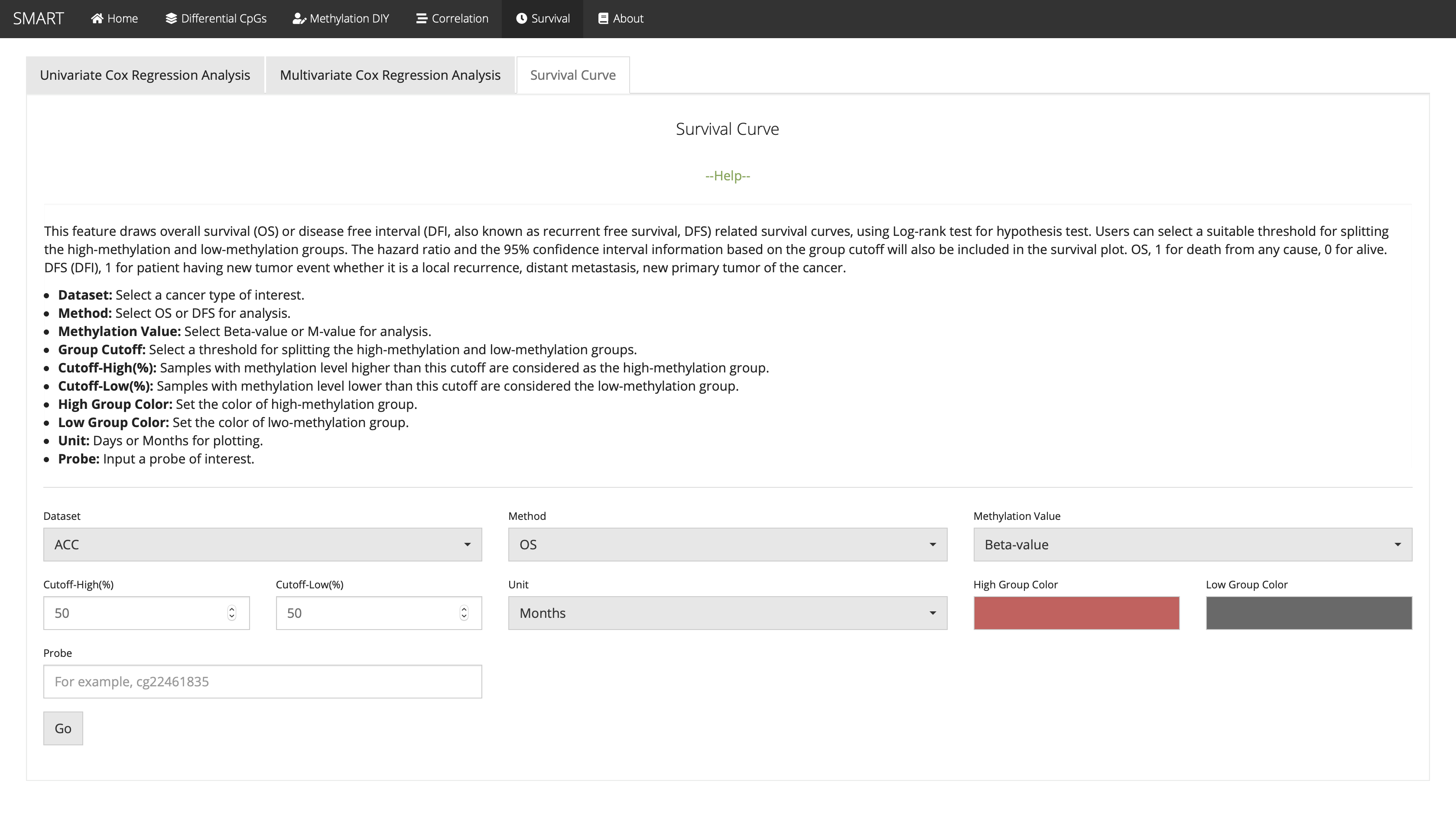
Survival Curve
This feature draws overall survival (OS) or disease free interval (DFI, also known as recurrent free survival, DFS) related survival curves, using Log-rank test for hypothesis test. Users can select a suitable threshold for splitting the high-methylation and low-methylation groups. The hazard ratio and the 95% confidence interval information based on the group cutoff will also be included in the survival plot. OS, 1 for death from any cause, 0 for alive. DFS (DFI), 1 for patient having new tumor event whether it is a local recurrence, distant metastasis, new primary tumor of the cancer.
About
Data
Getting Started
This is the SMART App start screen:
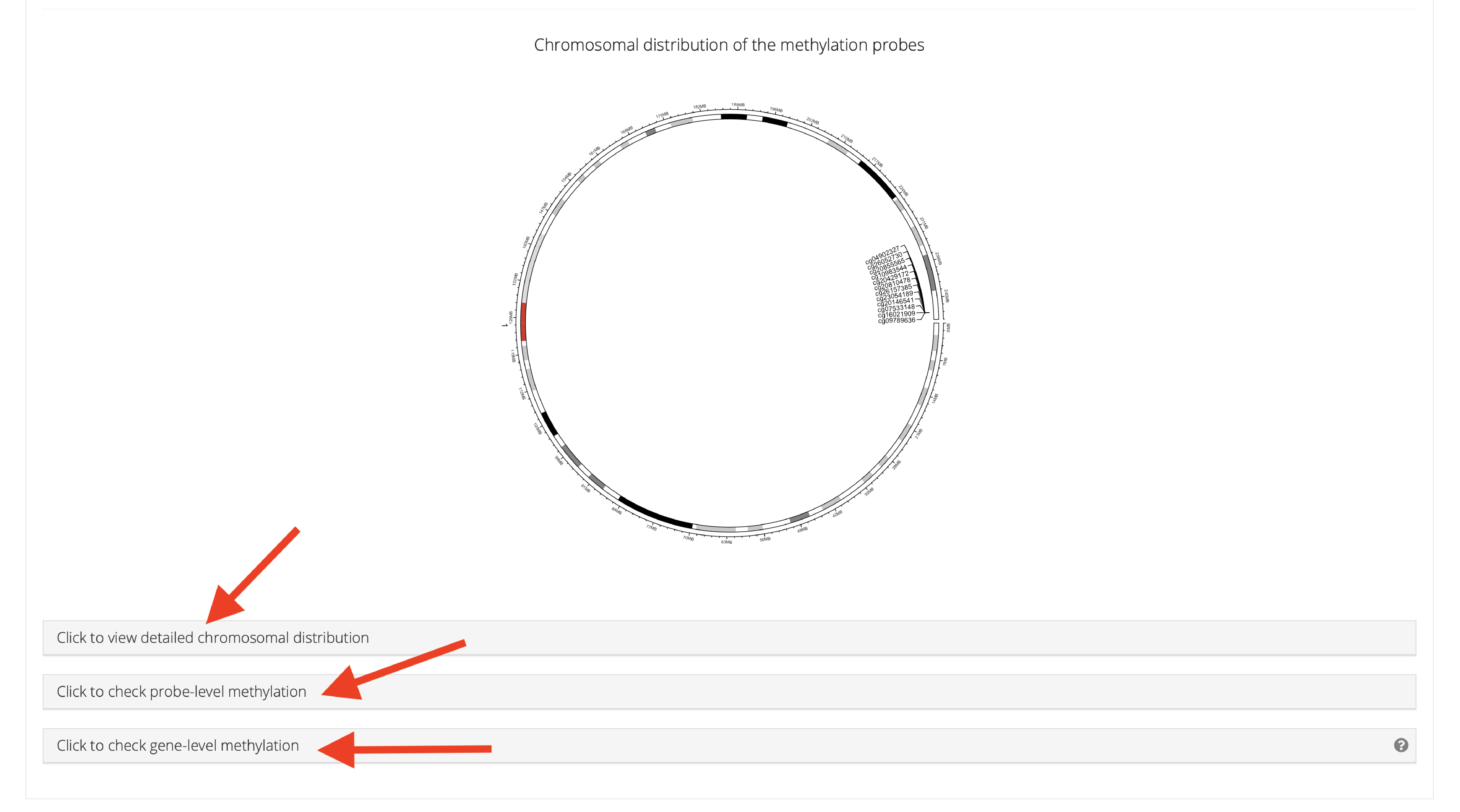
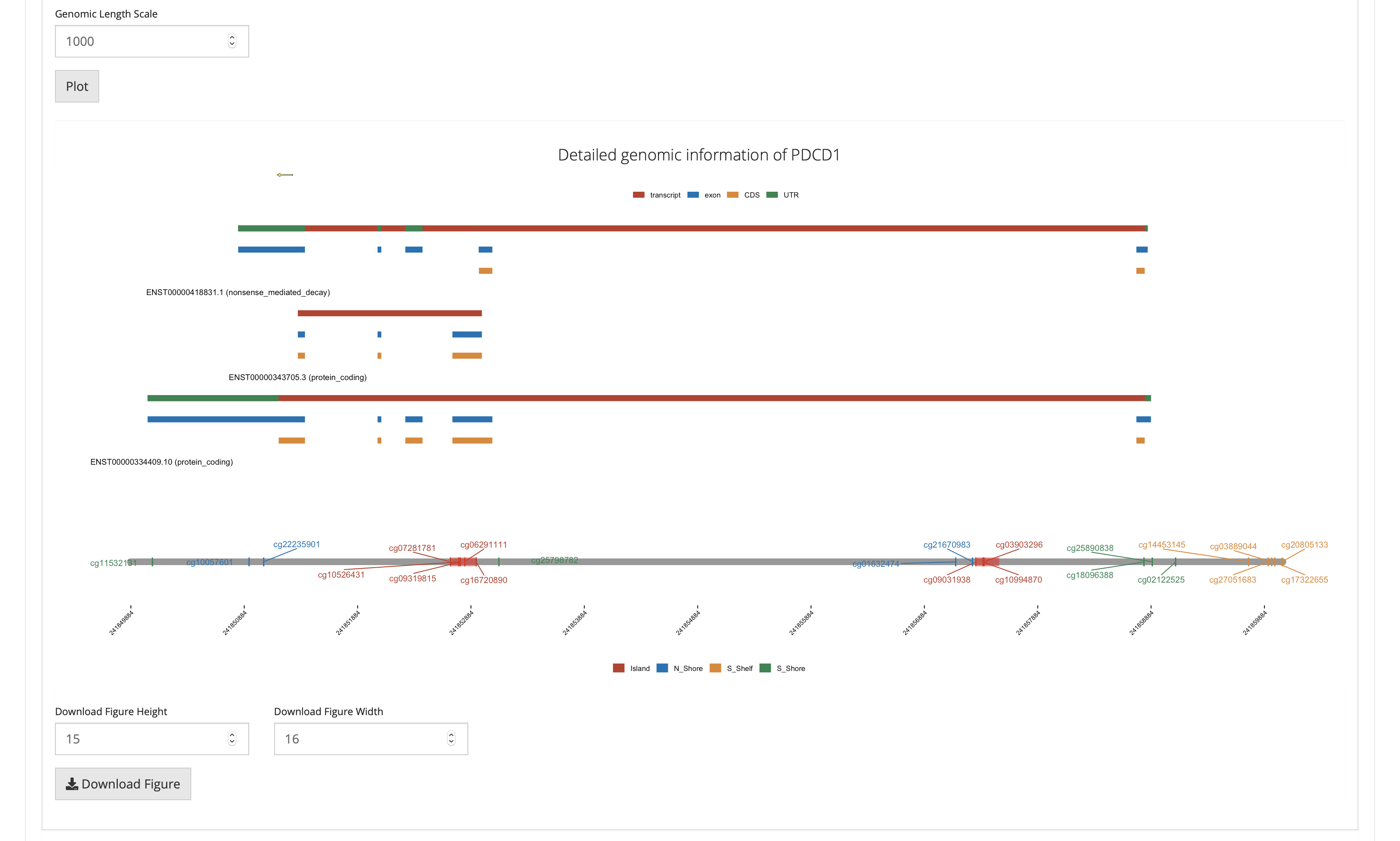
Differential analysis:
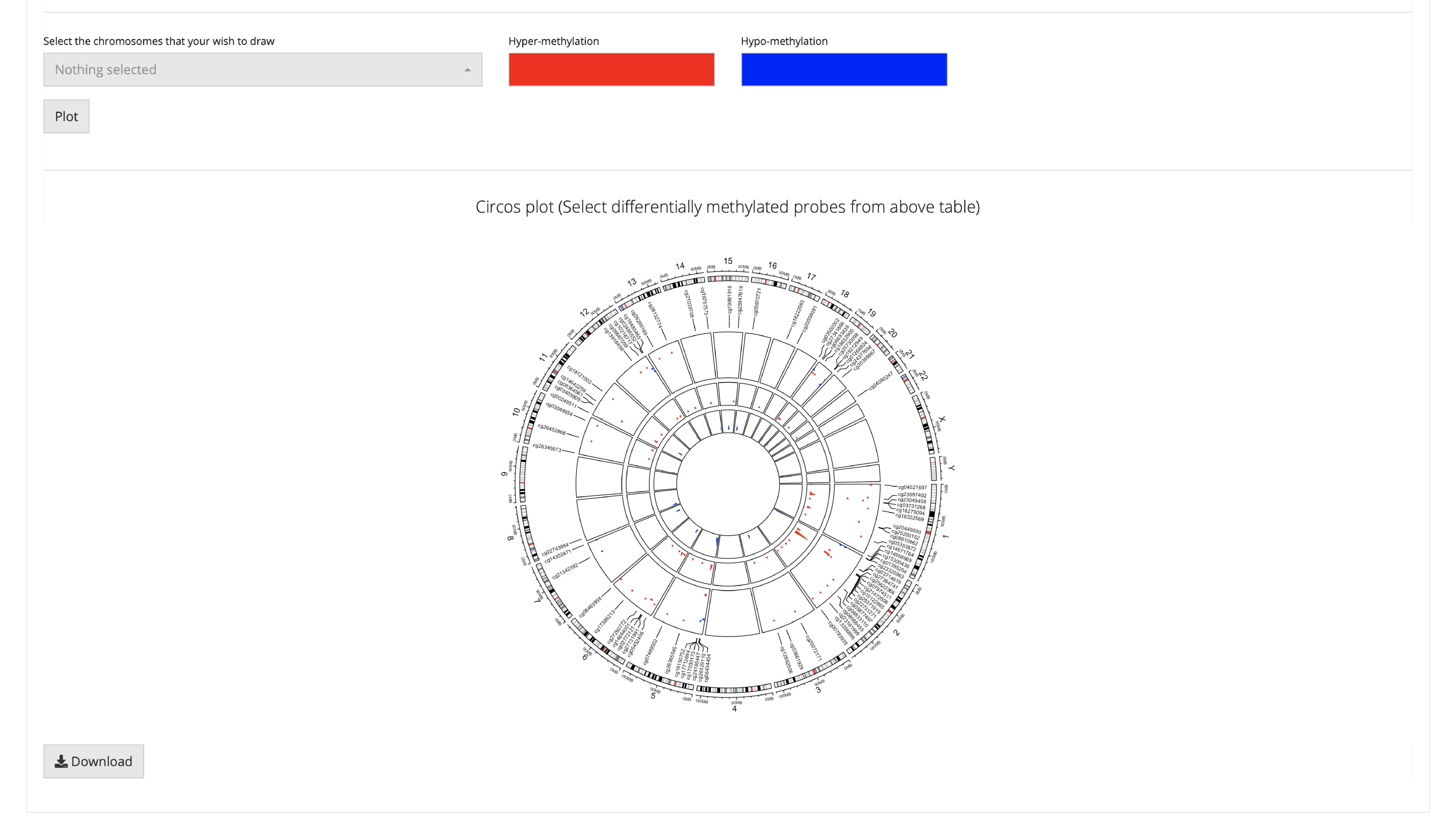
Methylation DIY:
Correlation:
Survival: